Opinion on Molecular Dynamics Simulations for Protein Design and Characterization
Posted: 2026-02-28
Here’s an opinion post about molecular dynamics simulations for protein design/characterization, something I’ve recently become rather passionate about! According to Hollingsworth and Dror (2018) in their review article “Molecular dynamics simulation for all” in Neuron (Ref), they note that the most intuitive reason to perform molecular dynamics simulations on biomolecules is to understand movement and flexibility, revealing their dynamic nature and relevant conformational changes that can lead to more information rich discoveries (like discoverying new drugs or understanding interactions).
Below, is figure 2 from Hollingsworth and Dror's review (Applications of molecular dynamics simulations):
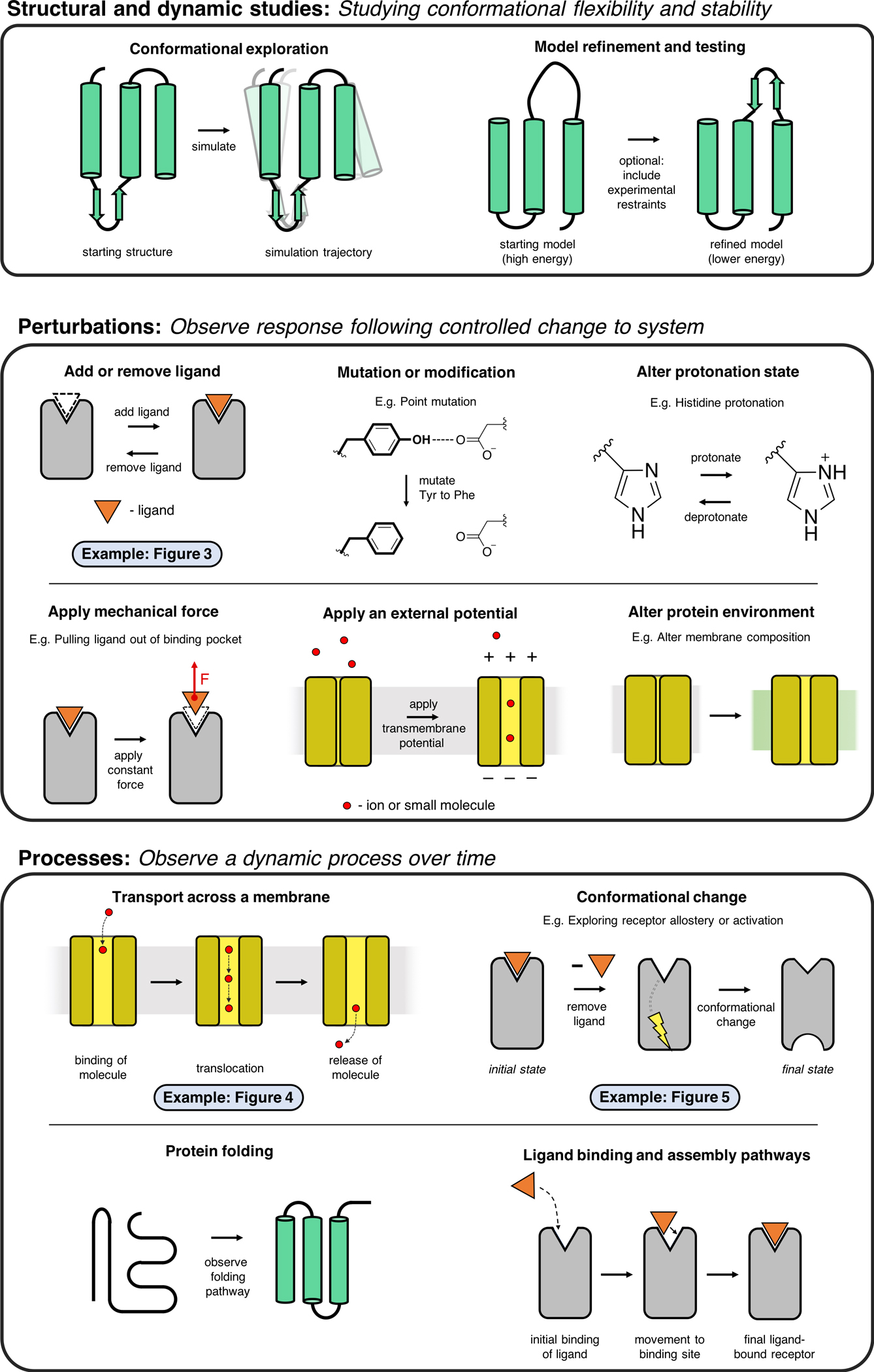
A exciting application is in drug discovery, where MD simulations can drive new wet lab experiments by testing biological questions computationally first. Many protein structures are averages and may have artifacts from crystal lattice packing or lack of a lipid bilayer, so MD simulations are utilized to refine structures to a more energy minimized form for more accurate downstream tasks. They can reveal the best binding pose for ligands and you can even subtract a ligand or mutate the protein binding site to see what effects that might have on a structure in a dynamic system, providing insights into disease or many biological questions. It's already proven useful for lead optimization for faster drug discovery by looking at the affinities for different ligands which generally performs better than docking methods.
MD simulations are not cheap. I’ve set up a typical simulation using the OpenMM Python library (Ref) to refine protein structures for a paper I’m writing on structure search. My pipeline is generally as follows (with a big shout out to Chase Harms for advising): fill in missing atoms like hydrogens —> solvate the system with the protein in water and ions, keeping a neutral charge —> run an energy minimization step to remove the significant steric clashes —> perform an NPT equilibration (which holds pressure and temperature constant) followed by a NVT equilibration with temperature ramping (holding volume constant) both keep alpha-carbons (part of the protein backbone) restrained to their position —> then a “production” run on the equilibrated protein without restraints to find a final energy minimized form. It can take up to 1 hour for one 200 amino acid protein on a single NVIDIA RTX 4090 GPU and even days for bigger proteins.
Molecular dynamics is not the only approach to studying or pertubating protein structures, but for refinement and understanding protein motion, it is a great approach (with the right setup and appropriate validation), making structures more biologically relevant. Check out the excellent review paper (link above) for even more applications. If you are doing any protein design, drug discovery, protein kinetics, interaction studies, or conformational change studies in biomolecules, it’s worth considering learning more about MD simulations on biomolecules. It is a dense field, combining chemistry, physics, biology and computing, but just may be worth the investment!